
The evaluation of the global medical devices market in 2023 was USD 518.46 billion and North America was a major player with a market share of ~40%. The global market is projected to flourish from USD 542.21 billion in 2024 to USD 886.80 billion by 2032. Further, market size in the United States (U.S.) alone is predicted to rise significantly, achieving a value of USD 314.96 billion [1].
Entry to this promising market in the U.S. is regulated by the Food & Drug Administration (FDA). Within the FDA, the Center for Biologics Evaluation and Research (CBER) regulates all tests (referred to as in vitro diagnostics) used for blood donor screening and medical devices used for blood collection and processing. All other medical devices fall under the purview of the Center for Devices and Radiological Health (CDRH). CDRH is responsible for regulating firms who manufacture, repackage, relabel, and/or import medical devices sold in the United States ensuring the safety and effectiveness of medical devices [2],[3],[4].
Before we dive into the details of classification, let’s first look at the definition of a medical device. Section 201(h) of the Federal Food, Drug, and Cosmetic Act broadly defines it as “any instrument, machine, contrivance, implant, in vitro reagent that’s intended to treat, cure, prevent, mitigate, diagnose disease in man”. Some examples could be a simple tongue depressor, or a thermometer, all the way to an advanced robotic surgical device.
Entering the U.S. market with a medical device involves a structured process that every manufacturer must navigate [5].

The journey begins with the crucial step of classifying the device according to its level of risk. This classification sets the foundation for determining the regulatory pathway and requirements necessary for gaining approval from the US FDA, ensuring compliance with stringent safety and effectiveness standards.
In this blog, we shall focus on the first crucial step of classification.
Medical devices are categorized into one of three classes (I, II, or III), based on the degree of risk they present. As the device class increases from class I to class II to class III, the regulatory controls also increase. The risk they present depend on material components, manufacturing processes and the clinical use of the device which includes intended anatomical location, frequency and duration of exposure [6].
The classes of devices, potential risk, and their examples are summarized in the following table [5].
|
Class |
Risk |
Potential Harm |
Examples |
|---|---|---|---|
|
I |
Lowest |
Present minimal potential for harm |
Tongue depressor, Toothbrush, Bandage, Oxygen mask, manual stethoscope, breathing mouthpiece, Hospital bed, non-electric wheelchair |
|
II |
Moderate |
Higher risk than Class I devices |
Wound dressing, Syringe, Catheter, Nebulizer, Oximeter, root canal filling resin, audiometer Pregnancy test kit, Contact lenses, Insulin pump |
|
III |
Highest |
Sustain or support life, are implanted, or present potential unreasonable risk of illness or injury |
Pacemaker, Breast implant, Ventilator, Heart valve, Defibrillators, Implanted prosthetics, Intraocular fluid, Foetal blood sampling monitors |
We shall discuss regulatory controls and pathways for approval of each class in the next blog. Let us now take a closer look at how to determine the appropriate class for your device.
How To Determine the Appropriate Class for Your Medical Device
Method 1: Search the Product Classification Database
You may use keywords to search the database to determine if there is an existing product classification applicable to your product using the link below.
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm
Here is one example of the keyword “wound dressing” searched in the database and the search results found for all 3 classes of devices:
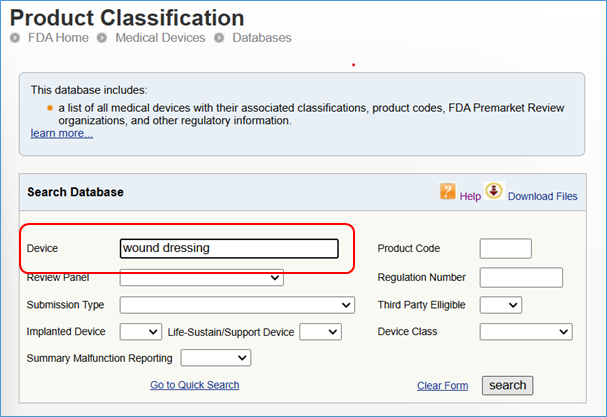
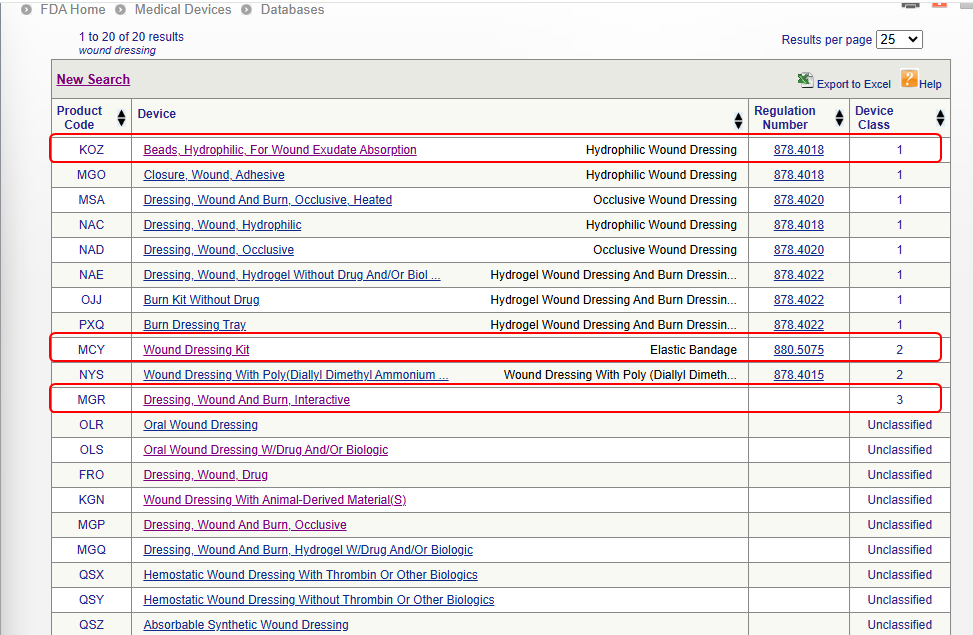
Method 2: Navigate the Device Panel
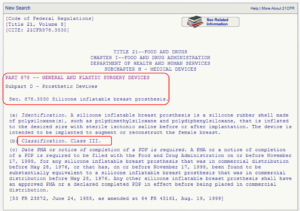
This option is for you if you know the medical speciality of your device. You can navigate through the device panel list: (https://www.fda.gov/medical-devices/classify-your-medical-device/device-classification-panels). This list is created by the FDA by organizing over 1700 types of devices in 16 categories/panels. These panels are found in Parts 862 – 892 in Title 21 of the Code of Federal Regulations (CFR). Here we show an example of the “General and Plastic Surgery” panel and representative device description of each class:
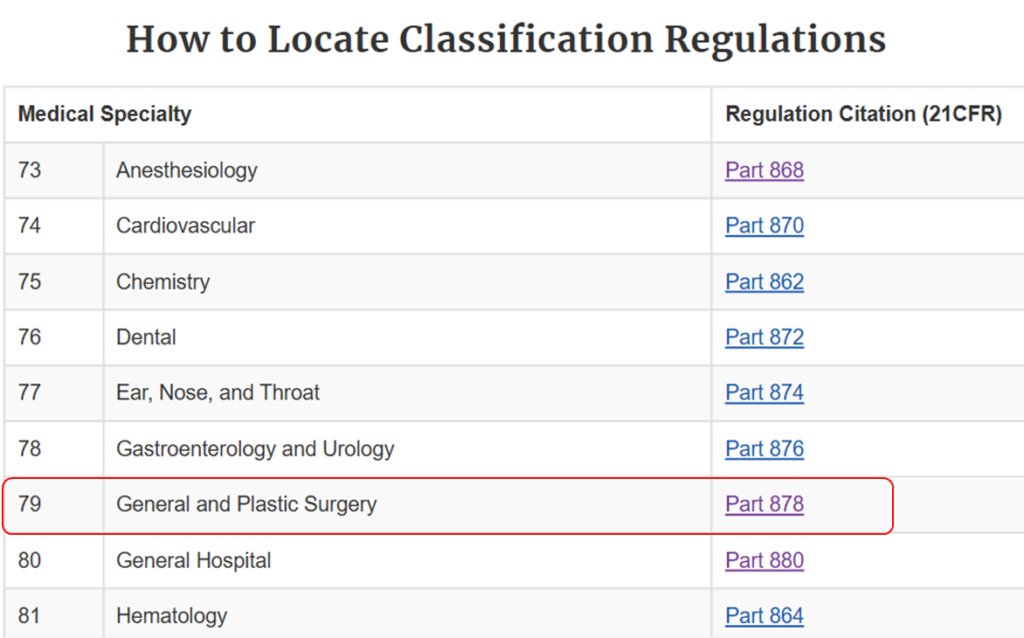
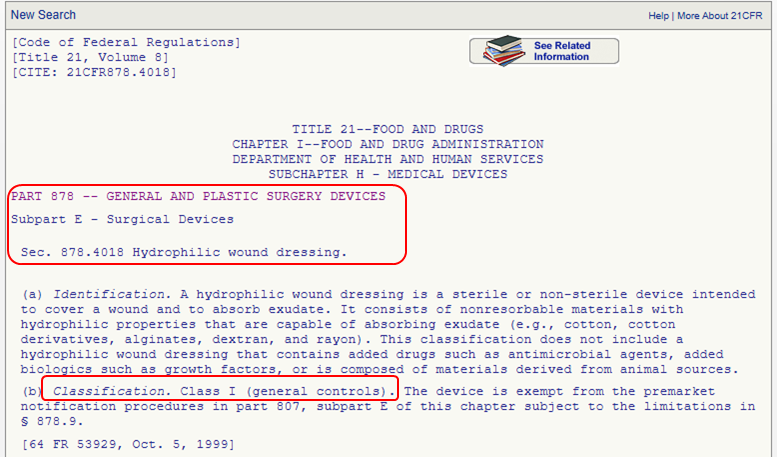
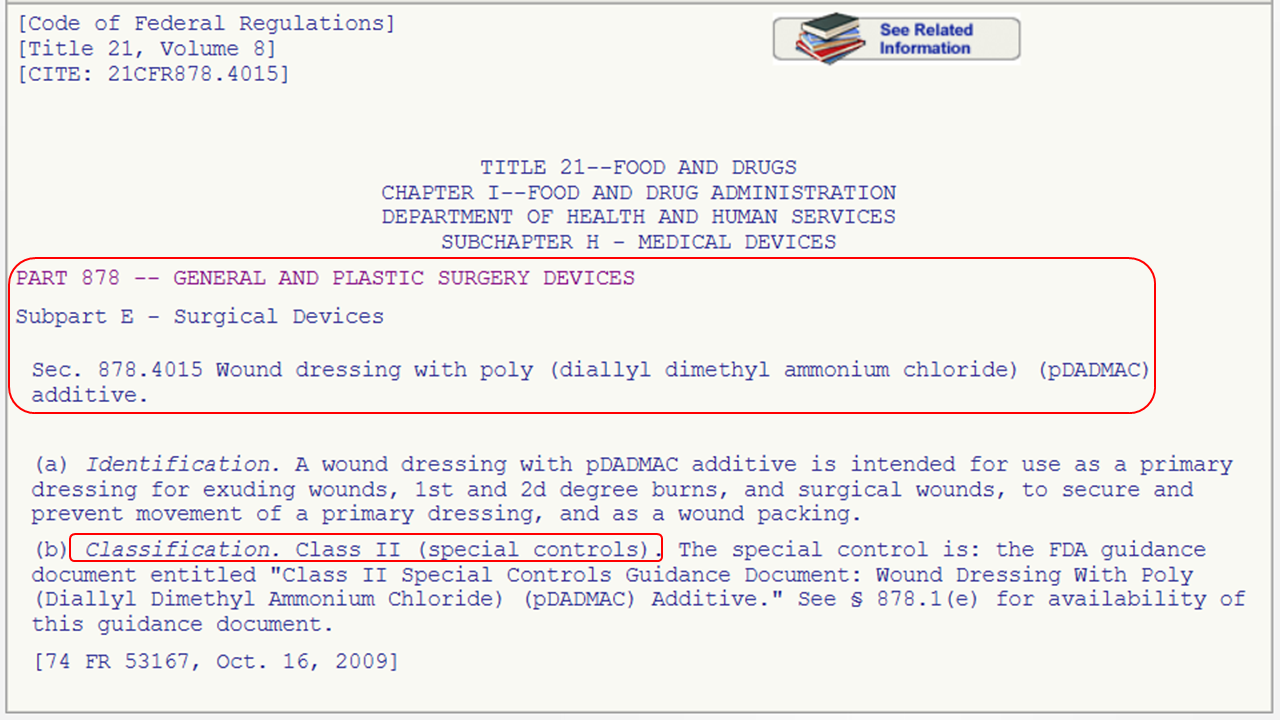
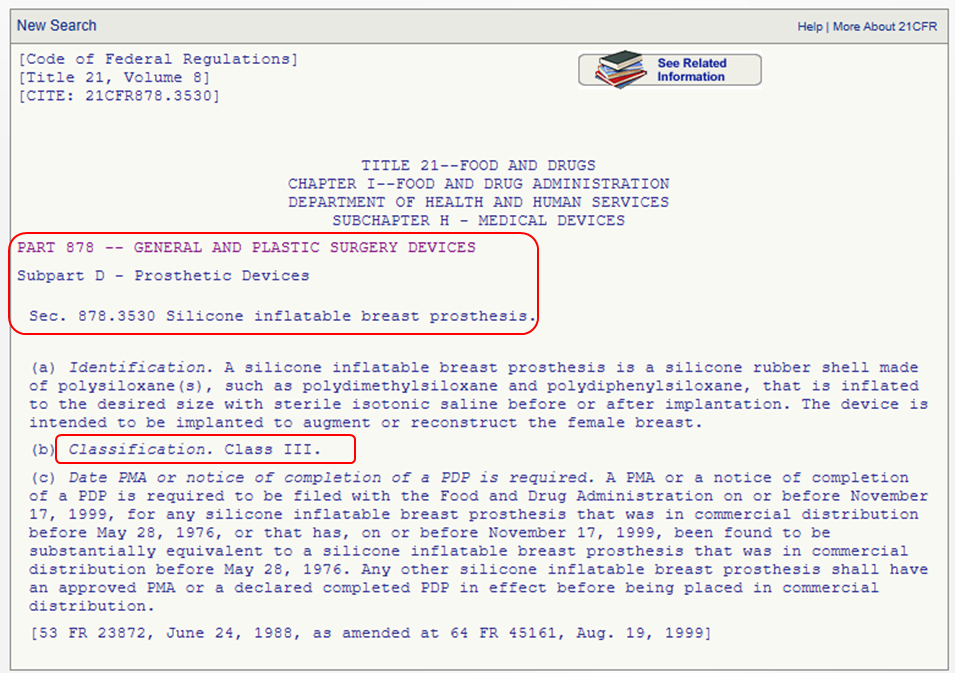
Method 3: 513(g) Request
If you would like a formal device determination or classification from the FDA, consider submitting a 513(g) Request. The FDA guidance document [7] and CDRH Learn training module [8] are helpful to submit this request.
Conclusion
Medical devices are spread in broad spectrum right from tongue depressors to pacemakers. US FDA classifies these medical devices into three main categories as Class I, II, and III based on the risk they present. Accordingly, the level of control necessary to assure their safety and effectiveness is regulated. We can take different steps to classify our medical device such as searching the classification database, navigating the panel list or submitting a formal request to the FDA. We shall deep dive into the regulatory controls required and pathways applicable for each class in the upcoming blog. Stay tuned!
Check out other informative blogs on our cutting-edge silk-based tissue engineering products here!
References
- https://www.fortunebusinessinsights.com/industry-reports/medical-devices-market-100085
- https://www.fda.gov/medical-devices/device-advice-comprehensive-regulatory-assistance/overview-device-regulation
- https://www.fda.gov/industry/fda-basics-industry/what-medical-devices-does-cber-regulate
- Principles and Practice of clinical Trial Medicine, ‘Chapter 2, Ethical, Legal and Regulatory Issues’, 2008, Pages 17-39
- https://www.fda.gov/medical-devices/device-advice-comprehensive-regulatory-assistance/how-study-and-market-your-device
- International Organization for Standardization. (2023). Biological evaluation of medical devices – Part 1: Evaluation and testing within a risk management process (ISO Standard No. 10993-1).
- FDA and Industry Procedures for Section 513(g) Requests for Information under the Federal Food, Drug, and Cosmetic Act Guidance (2012)
- https://fda.yorkcast.com/mediasite/Play/01b9a71491b54a2c9603f01443a477d61d

Author Bio – Dr. Arati Deshmukh is Manager – Regulatory at Serigen Mediproducts. She holds a Ph.D. in Biological Sciences from Academy of Scientific and Innovative Research, CSIR – National Chemical Laboratory. She has 5 years of professional experience in the biotherapeutics industry in regulatory science, analytical similarity assessment, data analysis and scientific writing.

The evaluation of the global medical devices market in 2023 was USD 518.46 billion and North America was a major player with a market share of ~40%. The global market is projected to flourish from USD 542.21 billion in 2024 to USD 886.80 billion by 2032. Further, market size in the United States (U.S.) alone is predicted to rise significantly, achieving a value of USD 314.96 billion [1].
Entry to this promising market in the U.S. is regulated by the Food & Drug Administration (FDA). Within the FDA, the Center for Biologics Evaluation and Research (CBER) regulates all tests (referred to as in vitro diagnostics) used for blood donor screening and medical devices used for blood collection and processing. All other medical devices fall under the purview of the Center for Devices and Radiological Health (CDRH). CDRH is responsible for regulating firms who manufacture, repackage, relabel, and/or import medical devices sold in the United States ensuring the safety and effectiveness of medical devices [2],[3],[4].
Before we dive into the details of classification, let’s first look at the definition of a medical device. Section 201(h) of the Federal Food, Drug, and Cosmetic Act broadly defines it as “any instrument, machine, contrivance, implant, in vitro reagent that’s intended to treat, cure, prevent, mitigate, diagnose disease in man”. Some examples could be a simple tongue depressor, or a thermometer, all the way to an advanced robotic surgical device.
Entering the U.S. market with a medical device involves a structured process that every manufacturer must navigate [5].

The journey begins with the crucial step of classifying the device according to its level of risk. This classification sets the foundation for determining the regulatory pathway and requirements necessary for gaining approval from the US FDA, ensuring compliance with stringent safety and effectiveness standards.
In this blog, we shall focus on the first crucial step of classification.
Medical devices are categorized into one of three classes (I, II, or III), based on the degree of risk they present. As the device class increases from class I to class II to class III, the regulatory controls also increase. The risk they present depend on material components, manufacturing processes and the clinical use of the device which includes intended anatomical location, frequency and duration of exposure [6].
The classes of devices, potential risk, and their examples are summarized in the following table [5].
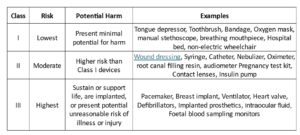
We shall discuss regulatory controls and pathways for approval of each class in the next blog. Let us now take a closer look at how to determine the appropriate class for your device.
How To Determine the Appropriate Class for Your Medical Device
Method 1: Search the Product Classification Database
You may use keywords to search the database to determine if there is an existing product classification applicable to your product using the link below.
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm
Here is one example of the keyword “wound dressing” searched in the database and the search results found for all 3 classes of devices:
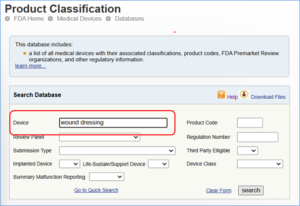
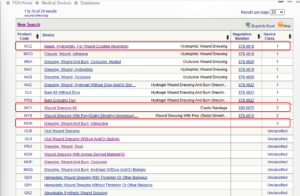
Method 2: Navigate the Device Panel
This option is for you if you know the medical speciality of your device. You can navigate through the device panel list:
(https://www.fda.gov/medical-devices/classify-your-medical-device/device-classification-panels).
This list is created by the FDA by organizing over 1700 types of devices in 16 categories/panels. These panels are found in Parts 862 – 892 in Title 21 of the Code of Federal Regulations (CFR). Here we show an example of the “General and Plastic Surgery” panel and representative device description of each class:
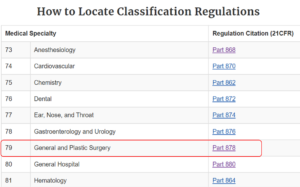
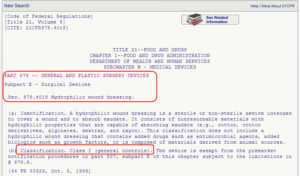
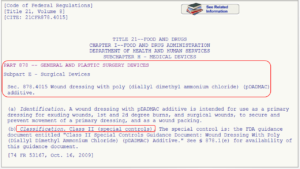
Method 3: 513(g) Request
If you would like a formal device determination or classification from the FDA, consider submitting a 513(g) Request. The FDA guidance document [7] and CDRH Learn training module [8] are helpful to submit this request.
Conclusion
Medical devices are spread in broad spectrum right from tongue depressors to pacemakers. US FDA classifies these medical devices into three main categories as Class I, II, and III based on the risk they present. Accordingly, the level of control necessary to assure their safety and effectiveness is regulated. We can take different steps to classify our medical device such as searching the classification database, navigating the panel list or submitting a formal request to the FDA. We shall deep dive into the regulatory controls required and pathways applicable for each class in the upcoming blog. Stay tuned!
Check out other informative blogs on our cutting-edge silk-based tissue engineering products here!
References
- https://www.fortunebusinessinsights.com/industry-reports/medical-devices-market-100085
- https://www.fda.gov/medical-devices/device-advice-comprehensive-regulatory-assistance/overview-device-regulation
- https://www.fda.gov/industry/fda-basics-industry/what-medical-devices-does-cber-regulate
- Principles and Practice of clinical Trial Medicine, ‘Chapter 2, Ethical, Legal and Regulatory Issues’, 2008, Pages 17-39
- https://www.fda.gov/medical-devices/device-advice-comprehensive-regulatory-assistance/how-study-and-market-your-device
- International Organization for Standardization. (2023). Biological evaluation of medical devices – Part 1: Evaluation and testing within a risk management process (ISO Standard No. 10993-1).
- FDA and Industry Procedures for Section 513(g) Requests for Information under the Federal Food, Drug, and Cosmetic Act Guidance (2012)
- https://fda.yorkcast.com/mediasite/Play/01b9a71491b54a2c9603f01443a477d61d
Author Bio –
Dr. Arati Deshmukh is Manager – Regulatory at Serigen Mediproducts. She holds a Ph.D. in Biological Sciences from Academy of Scientific and Innovative Research, CSIR – National Chemical Laboratory. She has 5 years of professional experience in the biotherapeutics industry in regulatory science, analytical similarity assessment, data analysis and scientific writing.










